by
,
Journal of Advanced Electronic Materials | Volume 2, Issue 1: 1-7, 2026 | DOI: 10.62762/JAEM.2026.936533
Abstract
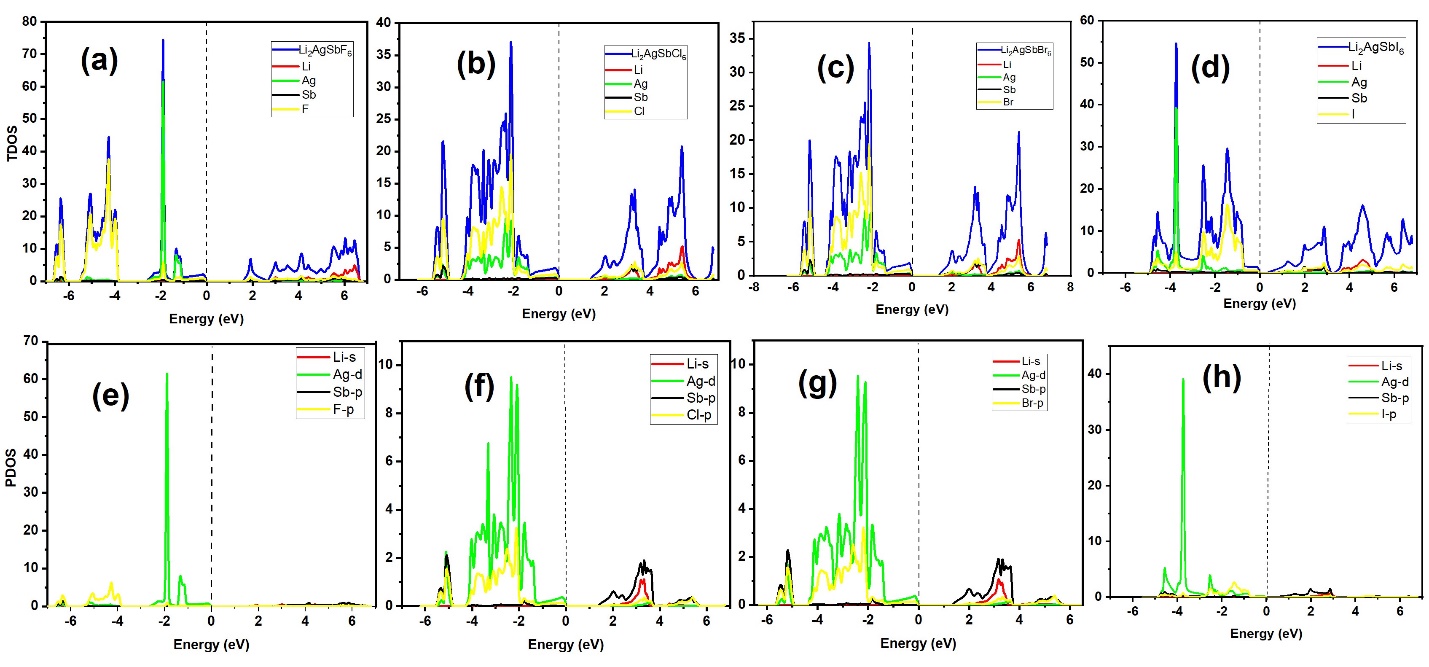
Halide double perovskites offer wide compositional flexibility and are being explored for energy-related applications. In this work, the structural, electronic, optical, and thermoelectric properties of Li\(_2\)AgSbX\(_6\) (X = F, Cl, Br, I) were investigated using density functional theory (DFT) within the WIEN2k package. Structural optimization shows a systematic increase in lattice parameter from F → I, accompanied by a decrease in bulk modulus, indicating higher compressibility for the heavier-halide compounds. Electronic-structure calculations within PBE-GGA identify all compositions as indirect-gap semiconductors, with band gaps decreasing across the series: 1.599 eV (F), 1.389 eV (C... More >
Graphical Abstract