Journal of Advanced Electronic Materials
ISSN: 3070-5649 (Online)
Email: [email protected]
 Submit Manuscript
Edit a Special Issue
Submit Manuscript
Edit a Special Issue

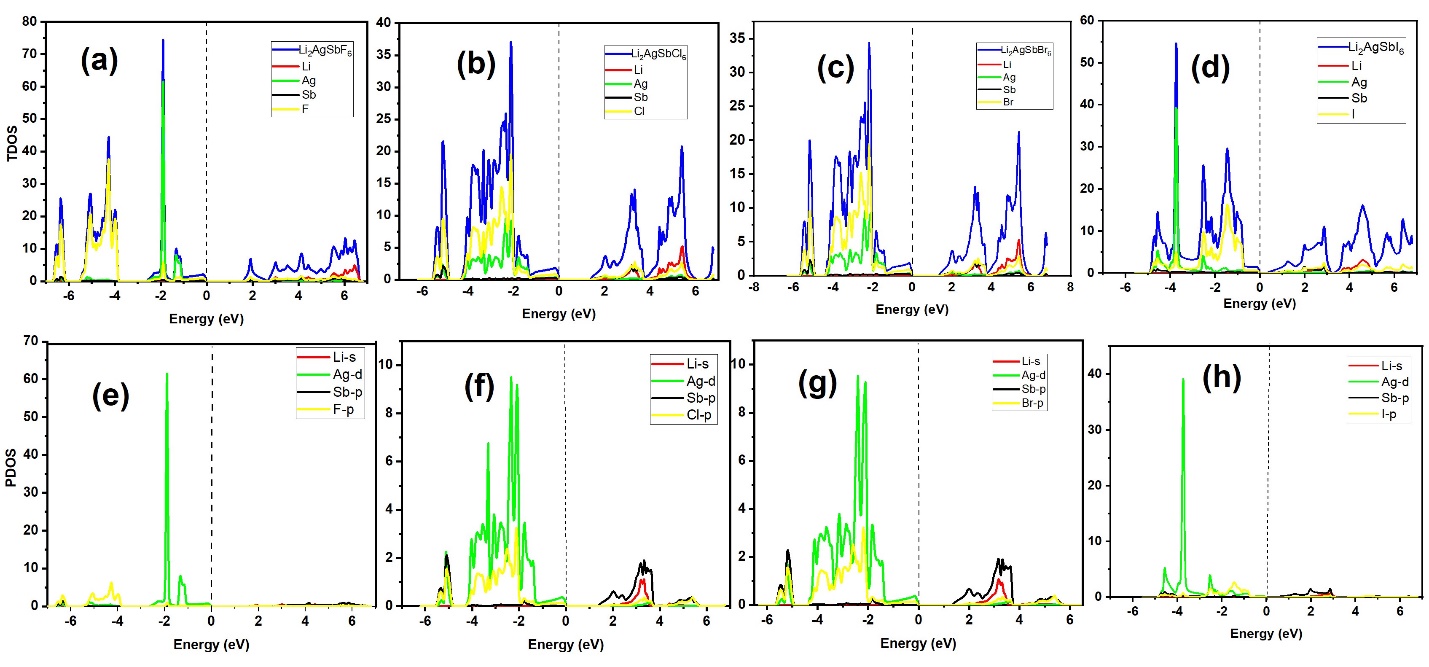
TY - JOUR AU - Gul, Seema AU - Sohail, Amir PY - 2026 DA - 2026/02/28 TI - Electronic Structure, Optical and Thermoelectric Properties of Li\(_2\)AgSbX\(_6\) (X = F, Cl, Br, I) Double Perovskites JO - Journal of Advanced Electronic Materials T2 - Journal of Advanced Electronic Materials JF - Journal of Advanced Electronic Materials VL - 2 IS - 1 SP - 1 EP - 7 DO - 10.62762/JAEM.2026.936533 UR - https://www.icck.org/article/abs/JAEM.2026.936533 KW - double perovskites KW - electronic properties KW - thermoelectric properties KW - optoelectronics AB - Halide double perovskites offer wide compositional flexibility and are being explored for energy-related applications. In this work, the structural, electronic, optical, and thermoelectric properties of Li\(_2\)AgSbX\(_6\) (X = F, Cl, Br, I) were investigated using density functional theory (DFT) within the WIEN2k package. Structural optimization shows a systematic increase in lattice parameter from F → I, accompanied by a decrease in bulk modulus, indicating higher compressibility for the heavier-halide compounds. Electronic-structure calculations within PBE-GGA identify all compositions as indirect-gap semiconductors, with band gaps decreasing across the series: 1.599 eV (F), 1.389 eV (Cl), 0.509 eV (Br), and 0.307 eV (I). Density-of-states analysis indicates that the valence band is dominated mainly by Ag and halogen states, while the conduction band is largely governed by Li and Sb contributions. Optical spectra derived from the calculated electronic structure (dielectric function and related optical constants) show strong optical activity and absorption spanning the visible-to-UV range, with the response shifting to lower photon energies for heavier halides. Thermoelectric transport coefficients were evaluated using BoltzTraP within the constant relaxation-time framework (\(\sigma / \tau\), \(\kappa_e / \tau\), S, and \(S^2 \sigma / \tau\)), and the reported ZT trend indicates that Li\(_2\)AgSbCl\(_6\) exhibits the most favorable and temperature-stable thermoelectric performance in the presented dataset (\(ZT \approx 1\)), whereas the I-based compound shows a pronounced reduction at elevated temperature. The Seebeck trends indicate p-type behavior for Li\(_2\)AgSbF\(_6\) and Li\(_2\)AgSbBr\(_6\) and n-type behavior for Li\(_2\)AgSbCl\(_6\) and Li\(_2\)AgSbI\(_6\) under the adopted transport conditions. SN - 3070-5649 PB - Institute of Central Computation and Knowledge LA - English ER -
@article{Gul2026Electronic,
author = {Seema Gul and Amir Sohail},
title = {Electronic Structure, Optical and Thermoelectric Properties of Li\(\_2\)AgSbX\(\_6\) (X = F, Cl, Br, I) Double Perovskites},
journal = {Journal of Advanced Electronic Materials},
year = {2026},
volume = {2},
number = {1},
pages = {1-7},
doi = {10.62762/JAEM.2026.936533},
url = {https://www.icck.org/article/abs/JAEM.2026.936533},
abstract = {Halide double perovskites offer wide compositional flexibility and are being explored for energy-related applications. In this work, the structural, electronic, optical, and thermoelectric properties of Li\(\_2\)AgSbX\(\_6\) (X = F, Cl, Br, I) were investigated using density functional theory (DFT) within the WIEN2k package. Structural optimization shows a systematic increase in lattice parameter from F → I, accompanied by a decrease in bulk modulus, indicating higher compressibility for the heavier-halide compounds. Electronic-structure calculations within PBE-GGA identify all compositions as indirect-gap semiconductors, with band gaps decreasing across the series: 1.599 eV (F), 1.389 eV (Cl), 0.509 eV (Br), and 0.307 eV (I). Density-of-states analysis indicates that the valence band is dominated mainly by Ag and halogen states, while the conduction band is largely governed by Li and Sb contributions. Optical spectra derived from the calculated electronic structure (dielectric function and related optical constants) show strong optical activity and absorption spanning the visible-to-UV range, with the response shifting to lower photon energies for heavier halides. Thermoelectric transport coefficients were evaluated using BoltzTraP within the constant relaxation-time framework (\(\sigma / \tau\), \(\kappa\_e / \tau\), S, and \(S^2 \sigma / \tau\)), and the reported ZT trend indicates that Li\(\_2\)AgSbCl\(\_6\) exhibits the most favorable and temperature-stable thermoelectric performance in the presented dataset (\(ZT \approx 1\)), whereas the I-based compound shows a pronounced reduction at elevated temperature. The Seebeck trends indicate p-type behavior for Li\(\_2\)AgSbF\(\_6\) and Li\(\_2\)AgSbBr\(\_6\) and n-type behavior for Li\(\_2\)AgSbCl\(\_6\) and Li\(\_2\)AgSbI\(\_6\) under the adopted transport conditions.},
keywords = {double perovskites, electronic properties, thermoelectric properties, optoelectronics},
issn = {3070-5649},
publisher = {Institute of Central Computation and Knowledge}
}
 Copyright © 2026 by the Author(s). Published by Institute of Central Computation and Knowledge. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
Copyright © 2026 by the Author(s). Published by Institute of Central Computation and Knowledge. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. 
Portico
All published articles are preserved here permanently:
https://www.portico.org/publishers/icck/